Genome annotation
Structural genome annotation
- Structural gene annotation - find out where the region of interest is
- Functional gene annotation - find out what the region do
- gff - genome feature file
Main steps:
QC assembly -> structural annotation -> manual curation -> functional annotation -> Submission or Downstream analysis
- QC of assembly is highly important
- Repeat Masking to improve the gene annotations
- most pipelines have this included, but check first before using a annotation program
- annotations are either based on proteins or transcripts
- if a protein is unknown then you won't annotate it
- RNA-seq tries to find all transcripts - should always be included in a annotation project
Approaches for annotations:
- Similarity-based methods - these use similarity to annotated sequences like proteins, cDNAs, or ESTs
- Ab initio prediction - likelihood based methods that needs to be trained (give them 1000 known genes of a species)
- annotations based on gene content (codon usage, GC content, exon/intron size, promotor, ORF, start codons, splice sites and more)
- sensitivity and specifity has to be determined after the training
- false positive results? overpredicting?
- sensitivity and specifity on nucleotide level is more important than on gene level when evaluating the performance
- Hybrid approaches - ab initio tools with the ability to integrate external evidence/hints
- Comparative (homology) based gene finders - these align genomic sequences from different species and use the alignments to guide the gene predictions
- Chooser, combiner approaches - these combine gene predictions of other gene finders
- Pipelines - These combine multiple approaches
- use a pipeline
Popular tools:
Supported by the maker tool:
- SNAP - Works ok, easy to train, not as good as others especially on longer intron genomes.
- Augustus - Works great, hard to train (but getting better)
- GeneMark-ES - Self training, no hints, buggy, not good for fragmented genomes or long introns (Best suited for Fungi).
- FGENESH - Works great, costs money even for training.
- GlimmerHMM (Eukaryote)
- GenScan
- Gnomon (NCBI)
- but you want to use those tools in a pipeline - combining various methodes since it improves highly the output quality
PIPELINES:
PASA
- Produces evidence-driven consensus gene models
- (-) minimalist pipeline
- (+) good for detecting isoforms
- (+) biologically relevant predictions
- using Ab initio tools and combined with EVM it does a pretty good job
- PASA + Ab initio + EVM is not automatized
NCBI pipeline
- best one yet - but difficult to install
- NCBI staff can be asked an they help you
- Evidence + ab initio (Gnomon), repeat masking, gene naming, data formatting, miRNAs, tRNAs
Ensembl
- Evidence based only ( comparative + homology )
MAKER2
- Evidence based and/or ab initio
- developed as an easy-to-use alternative to other pipelines
- Easy to use and to configure
- Almost unlimited parallelism built-in (limited by data and hardware)
- Largely independent from the underlying system it is run on
- Everything is run through one command, no manual combining of data/outputs
- Follows common standards, produces GMOD compliant output
- Annotation Edit Distance (AED) metric for improved quality control
- Provides a mechanism to train and retrain ab initio gene predictors
- Annotations can be updated by re-launching Maker with new evidence
- Pipelines give good results, MAKER2 the most flexible, adjustable
- Most methods only build gene models, no functional inference
- Computational pipelines make mistakes
- Annotation requires manual curation
- As for assembly, an annotation is never finished, it can always be improved (e.g. Human)
Genome annotation with augustus - practical
- we are using chromosome 4 of the fruit fly, Drosophila melanogaster
- QC of the assembly first:
- Fragmentation? (N50, N90, how many short contigs)
- Sanity of the fasta file (Presence of Ns, presence of ambiguous nucleotides, presence of lowercase nucleotides, single line sequences vs multiline sequences)
- completeness (using BUSCO)
- presence of organelles
- Others (GC content, How distant the investigated species is from the others annotated species available)
1. BUSCO for Assembly check
- BUSCO provides quantitative measures for the assessment of genome assembly, gene set, and transcriptome completeness, based on evolutionarily-informed expectations of gene content from near-universal single-copy orthologs selected from OrthoDB v9.
- first get the best dataset from the busco website
wget http://busco.ezlab.org/datasets/metazoa_odb9.tar.gz
tar xzvf metazoa_odb9.tar.gz
- launching BUSCO, to create the folder 4_dmel_busco
- inside is a
short_summary_4_dmel_busco.txtfor statistics on how many genes/regions could be found in comparision to the reference genome - the better the assembly the more will be found
BUSCO.py -i ~/annotation_course/data/genome/4.fa -o 4_dmel_busco -m geno -c 8 -l metazoa_odb9
fasta_statisticsAndPlot.plis used for additional statistics- Part of this github rep called GAAS
fasta_statisticsAndPlot.pl -f ~/annotation_course/data/genome/4.fa
2. augustus an ab initio gene finder
- if satisfied by the quality of the assembly we start the annotation
- we focus on the gene finder
augustus - these gene finders use likelihoods to find the most likely genes in the genome
- they are aware of start and stop codons and splice sites, and will only try to predict genes that follow these rules
- the most important factor here is that the gene finder needs to be trained on the organism you are running the program on, otherwise the probabilities for introns, exons, etc. will not be correct
- we have training files for Drosophila
Augustus * call it with a genome/assembly and it saves the annotation as a gff3 file
augustus --species=fly ~/annotation_course/data/genome/4.fa --gff3=yes > augustus_drosophila.gff
#to get additional isoforms use this:
augustus --species=fly ~/annotation_course/data/genome/4.fa --gff3=yes --alternatives-from-sampling=true > augustus_drosophila_isoform.gff
gff3_sp_statistics.plis also from GAAS for the created gff file fromaugustus- gives an statistical overview of the gene annotations
gff3_sp_statistics.pl --gff augustus_drosophila.gff
3. Visualisation
- opened the chromosome 4 file and the gff file in UGENE
- looks like this:

- An other good and popular software to explore genomes is IGV
maker gene build pipeline - practical
MAKER is a computational pipeline to automatically generate annotations from a range of input data. The Maker pipeline can work with any combination of the following data sets, which are put into the maker_opts.ctl:
- Proteins from the same species or related
- Swissport for high quality protein sequences
- Refseq sequence sets from the ftp-servers
- Ensembl using Biomart interface to download data for a specific region or a specific gene
- Proteins from more distantly related organisms
- Uniprot to include protein sequences from organisms closely related to your study organism
- EST sequences from the same species or very closely related species
- NCBI or EBI websites to retrieve such kind of data
- RNA-seq data from the same or very closely related species, in the form of splice sites or assembled transcripts
- normally genereted by yourself or are retrived on the Sequence Read Archive of NCBI (SRA) or the ENA of EBI
- ab initio predictions from one or more tools (supported are: Augustus, Snap, GeneMark, Fgenesh)
1. Start maker
- do
maker -CTLto create the 3 files for the pipeline (maker_opts.ctl, maker_bopts.ctl, maker_exe.ctl)
2.1 Evidence-based annotation
- the best protein- and EST-alignments are chosen to build the most likely gene model, without an ab initio model
We need to prepare various files and add their path to the
maker_opts.ctlfile (use , to seperate files):
- name of the genome sequence (
genome=)- name of the 'EST' set file(s) (
est=)- name of the 'Protein' set file(s) (
protein=)- name of the repeatmasker and repeatrunner files (
rm_gff=)- disabling ab initio with
protein2genome=1,est2genome=1- we deactivated the parameters
model_org=andrepeat_protein=to avoid the heavy work of repeatmasker (blank values)
- if the
maker_opts.ctlis configured properly start the maker pipeline with
mpiexec -n 8 maker # -n is core numbers
# and compile the output with:
maker_merge_outputs_from_datastore.pl --output maker_no_abinitio
- in the folder you find the annotation file
maker.gff - to get statistics use the
gff3_sp_statistics.pl
gff3_sp_statistics.pl --gff maker_no_abinitio/annotationByType/maker.gff
2.2 Run Maker with ab-initio predictions
- builds on the gff from step 2.1 and does a ab-initio prediction
- These files were present(repeatmasker.chr4.gff, repeatrunner.chr4.gff, 4.fa)
- These were created in Step 2.1 (est_gff_stringtie.gff, est2genome.gff, protein2genome.gff)
- so add them to the
maker_opts.ctl - change these now to
protein2genome=0,est2genome=0,keep_preds=1,augustus_species=fly - With these settings, Maker will run augustus to predict gene loci, but inform these predictions with information from the protein and est alignments
- You can combine step 2.1 & 2.2 into one
- its basically step 2.1 with augustus and ab initio activated (the gffs are created during the pipeline)
mpiexec -n 8 maker
#compiling
maker_merge_outputs_from_datastore.pl --output maker_with_abinitio
#check statistics with this GAAS script
gff3_sp_statistics.pl --gff maker_with_abinitio/annotationByType/maker.gff
Summary: We created 2 maker.gff files. One in maker_with_abinto/ one in maker_noabinto directory/. Doing Step 2.2 helped to make the gene predictions less fragmented. For best performance go all th way to step 2.2.
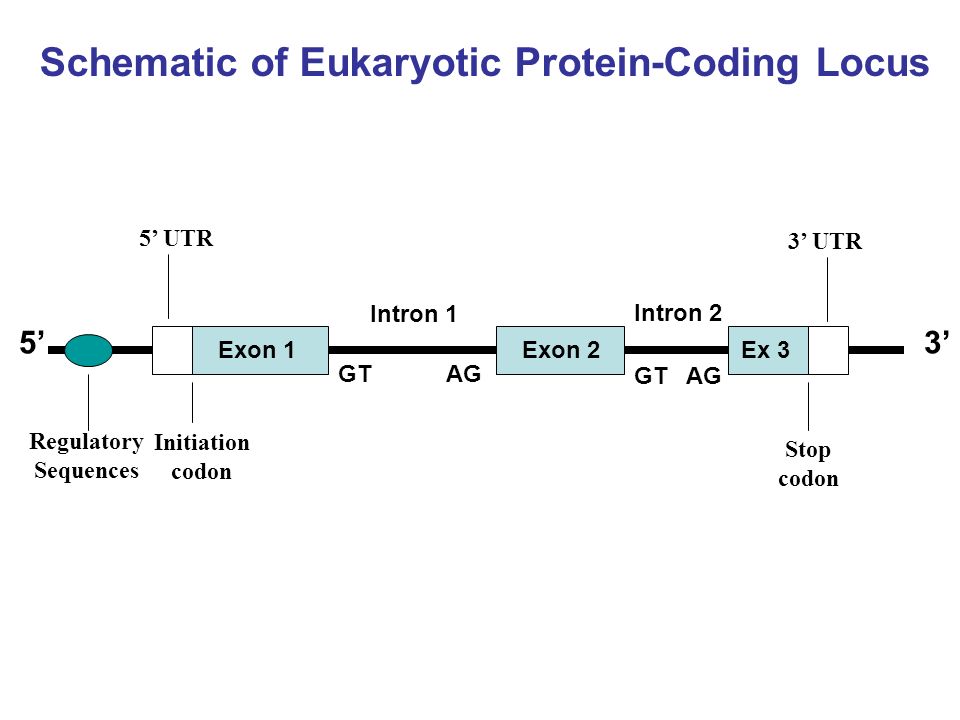
For Intron / Exon manual curation look for the GT/AG in an Intron 5'-3' [EXON]++GT--intron--AG++[EXON] 3'-5' [EXON]++GA--intron--TG++[EXON]
Functional genome annotation
- Functional gene annotation - find out what the region does
- You can do this experimentally (slow and expensive) or computationally
Computationally: Sequence based - mainly done * based on similarity/motif/profile * orthology based on evolutionary relationship * Clustering with KOG/COG * Synteny: Satsuma + kraken + custom script * phylogeny based Structure based * global structure comparison * localized regions * active sites resides * Protein-Protein Interaction data
blast based functional genome annotation
- you need Sequence, gff3 files
- Uniprot (exhaustive) or Swissprot (reliable)
- Use Annie to extract best hits from blast-hit list and the corresponding description from uniprot-headers
- Add the information to the annotation.gff using custom-script
- then you have a nice description of the blast hit in the gff file
| Database | Information | Comment |
|---|---|---|
| KEGG | Pathway | Kyoto Encyclopedia of Genes and Genomes |
| MetaCyc | Pathway | Curated database of experimentally elucidated metabolic pathways from all domains of life (NIH) |
| Reactome | Pathway | Curated and peer reviewed pathway database |
| UniPathway | Pathway | Manually curated resource of enzyme-catalyzed and spontaneous chemical reactions. |
| GO | Gene Ontology | Three structured, controlled vocabularies (ontologies) : biological processes, cellular components and molecular functions |
| Pfam | Protein families | Multiple sequence alignments and hidden Markov models |
| Interpro | P. fam., domains & functional sites | Run separate search applications, and create a signature to search against Interpro. |
| Tool | Approach | Comment |
|---|---|---|
| Trinotate | Best blast hit, protein domain identification (HMMER/PFAM), protein signal peptide and transmembrane domain prediction (signalP/tmHMM), and leveraging various annotation databases (eggNOG/GO/Kegg databases). | Not automated |
| Annocript | Best blast hit | Collects the best-hit and related annotations (proteins, domains, GO terms, Enzymes, pathways, short) |
| Annot8r | Best blast hits | A tool for Gene Ontology, KEGG biochemical pathways and Enzyme Commission EC number annotation of nucleotide and peptide sequences. |
| Sma3s | Best blast hit, Best reciprocal blast hit, clusterisation | 3 annotation levels |
| afterParty | BLAST, InterProScan | web application |
| Interproscan | Separate search applications, HMMs, fingerprints, patterns of InterPro | Created to unite secondary databases |
| Blast2Go | get best blast hits | Retrieve only GO,Commercial ! |
Interproscan approach - practical
- we took a gff file and the corresponding fasta file
- we had a pre downloaded database
- we used this script to extract the protein sequences with an gff annotation (
AA.fa)
gff3_sp_extract_sequences.pl --gff maker_with_abinitio.gff -f 4.fa -p --cfs -o AA.fa
- we used the interproscan complete thing - incl. databases (48 GB) to get the information
- there is a way to analyse through the web but it has a limit on requests
#takes 2-3 seconds per protein
interproscan.sh -i AA.fa -t p -dp -pa -appl Pfam,ProDom-2006.1,SuperFamily-1.75 --goterms --iprlookup
- load the retrieved functional information in your annotation file writing your own script or use the
makerscript
ipr_update_gff maker_with_abinitio.gff AA.fa.tsv > maker_with_abinitio_with_interpro.gff
BLAST approach - practical
- similar to your resistance gene blasts
blastp -db ~/annotation_course/data/blastdb/uniprot_dmel/uniprot_dmel.fa -query AA.fa -outfmt 6 -out blast.out -num_threads 8
- we are using
annieto process the blast output
git clone https://github.com/genomeannotation/Annie.git
# This is the annie.py script
Annie/annie.py -b blast.out -db ~/annotation_course/data/blastdb/uniprot_dmel/uniprot_dmel.fa -g maker_with_abinitio.gff -o annotation_blast.annie
-
Annie writes in a 3-column table format file, providing gene name and mRNA product information. The purpose of annie is relatively simple. It recovers the information in the sequence header of the uniprot fasta file, from the best sequence found by Blast (the lowest e-value).
-
load the retrieved functional information in your annotation file writing your own script or use the
makerscript
maker_gff3manager_JD_v8.pl -f maker_with_abinitio_with_interpro.gff -b annotation_blast.annie --ID FLY -o finalOutputDir
- we change the [product] tag to the [description] tag so Webapollo shows the actual protein name directly in the screen instead of just hovering over it
/home/student/.local/GAAS/annotation/WebApollo/gff3_webApollo_compliant.pl --gff finalOutputDir/codingGeneFeatures.gff -o final_annotation.gff
Subbmitting to EBI using a tool
In order to submit to EBI, the use of a tool like EMBLmyGFF3 will be your best choice.
Let's prepare your annotation to submit to ENA (EBI) You need to create an account and create a project asking a locus_tag for your annotation. You have to fill lot of metada information related to the assembly and so on. We will skip those tasks using fake information. First you need to download and install EMBLmyGFF3:
pip install --user git+https://github.com/NBISweden/EMBLmyGFF3.git
EMBLmyGFF3 finalOutputDir/codingGeneFeatures.gff 4.fa -o my_annotation_ready_to_submit.embl
You now have a EMBL flat file ready to submit.